What are the Causes of Pulmonary Arterial Hypertension?
While we do not fully understand what causes pulmonary arterial hypertension, studies have identified certain risk factors, diseases, and substances associated with developing PAH. Experts hope to continue to learn more about the specific causes of pulmonary hypertension as they continue to research the disease.
Idiopathic Pulmonary Arterial Hypertension:
Primary pulmonary hypertension or PPH is an old term no longer used that describes pulmonary arterial hypertension that cannot directly be explained by other diseases or exposures to substances. Today we use the term idiopathic pulmonary arterial hypertension to describe PH that has no other explanation. IPAH means that the elevated pulmonary arterial pressures cannot be explained by left heart disease, lung disease, sleep apnea, congenital heart disease, connective tissue disease, HIV, or left heart problems.
 Hereditary (or familial) Pulmonary Arterial Hypertension:
Hereditary (or familial) Pulmonary Arterial Hypertension:
PAH can be hereditary or run in families. A gene called BMPR2 has been linked to familial pulmonary hypertension. Although the inheritence is autosomal dominant (one defective gene copy is enough to cause disease), only about 20% of patients with a bad gene copy develop the disease. This is called incomplete penetrence. Early and routine screening is important for families with known PAH as the disease tends to affect each additional generation at an earlier age and often times can be more severe with each generation. Echocardiograms are the screening tool of choice for patients with familial pulmonary hypertension in their families as they are noninvasive and readily available.
Pulmonary Hypertension Related to Congenital Heart Disease:
Congenital heart diseases are those that are present at birth and involve abnormalities with the structure of the heart and the blood vessels of the lungs. Many forms of congenital heart 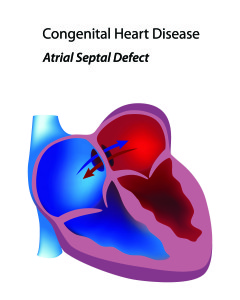 disease can lead to pulmonary arterial hypertension. Some patients will know of the defect in their heart from the time they are born and others may not find out until they are being evaluated for pulmonary hypertension. One of the more common congenital heart abnormalities associated with pulmonary arterial hypertension is an ASD or atrial septal defect. An ASD is a hole in the heart allowing blood to flow between the top two chambers (atria) of the heart. Depending on the size of the hole, extra blood flow that passes through the pulmonary arteries can lead to the changes of PAH. The larger the hole, the greater the risk of PAH. Small holes rarely lead to PAH. As the pressure in the pulmonary arteries increases, more blue (deoxygenated) blood from the right atrium is pushed into the left atrium without going through the lung circulation and picking up oxygen. These patients tend to do very well on pulmonary arterial hypertension therapies as the hole acts like a pop off valve to release pressure on the right side of the heart. It is very important that these patients be seen by pulmonary hypertension experts as physicians less aware of PAH may want to surgically close the ASD, a procedure that could be deadly in a PAH patient.
disease can lead to pulmonary arterial hypertension. Some patients will know of the defect in their heart from the time they are born and others may not find out until they are being evaluated for pulmonary hypertension. One of the more common congenital heart abnormalities associated with pulmonary arterial hypertension is an ASD or atrial septal defect. An ASD is a hole in the heart allowing blood to flow between the top two chambers (atria) of the heart. Depending on the size of the hole, extra blood flow that passes through the pulmonary arteries can lead to the changes of PAH. The larger the hole, the greater the risk of PAH. Small holes rarely lead to PAH. As the pressure in the pulmonary arteries increases, more blue (deoxygenated) blood from the right atrium is pushed into the left atrium without going through the lung circulation and picking up oxygen. These patients tend to do very well on pulmonary arterial hypertension therapies as the hole acts like a pop off valve to release pressure on the right side of the heart. It is very important that these patients be seen by pulmonary hypertension experts as physicians less aware of PAH may want to surgically close the ASD, a procedure that could be deadly in a PAH patient.
Pulmonary Hypertension Related to Connective Tissue Disease:
Patients with connective tissue diseases such as scleroderma also have an increased risk of developing PAH. These patients are often screened for pulmonary hypertension with annual echocardiograms and are referred to pulmonary hypertension experts immediately if any other symptoms of PAH present such as shortness of breath, poor exercise tolerance, or dizziness or fainting. PH patients with connective tissue diseases are treated very aggressively with pulmonary hypertension therapies because they progress quickly from mild to severe PAH. It is not fully understood why these patients do not respond to treatment as well as other groups of PH patients. Other connective tissue diseases that are associated with PAH include Lupus and Mixed Connective Tissue Disease. Rarely, Rheumatoid Arthritis is complicated by PAH.
Pulmonary Hypertension Related to Drugs or Toxins:
Patients that abuse or have abused methamphetamines or cocaine also have an increased risk of developing pulmonary arterial hypertension. Every patient being evaluated for pulmonary hypertension will be asked if they currently or have ever used any type of drugs in the past. It is important for the patient to be honest with their health care team so they can appropriately assess the risks someone has of developing PAH. If a patient is currently addicted to illegal substances their physician may be able to help them connect with resources to help them fight their addiction. The most important treatment for pulmonary hypertension related to drugs or toxins is to stop the drug or toxin. Certain diet pills such as Fen-Phen have also been linked to developing PAH. This group of patients will also be treated with therapies approved for PAH.
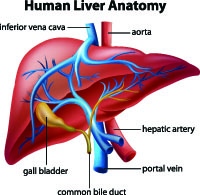 Portopulmonary Hypertension:
Portopulmonary Hypertension:
Patients with advanced liver disease may also develop pulmonary arterial hypertension (PAH). All patients being evaluated for liver transplantation will be evaluated for pulmonary hypertension. It is important to diagnose and treat PAH in this group of patients as transplantation requirements demand that a patient’s mean pulmonary artery pressure be less than or equal to 35mm Hg. These patients are generally treated very aggressively with oral and prostanoid therapies as soon as the diagnosis is made with a goal of making them a candidate for transplantation. Close monitoring is important while initiating therapies for this group of patients as their bodies metabolize the drugs differently and they may require less medication than other pulmonary hypertension patients. The liver transplant team will want repeat right heart catheterizations over time to document the mean PA (pulmonary artery) pressure. When a portopulmonary hypertension patient receives a liver transplant, continuous prostanoid therapy can often be discontinued immediately. The patient may or may not need to continue on oral therapies. Certain medications are avoided and others used with increased caution in patients with advanced liver disease.